Información para prescribir Bridion®
BRIDION® Solución inyectable, 100 mg/mL
PC-MK8616-EMEA/H/C/000885/II/0042-092020
1. DENOMINACIÓN DEL PRODUCTO MEDICINAL
BRIDION® 100 mg/mL solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
1 mL contiene sugammadex sódico equivalente a 100 mg de sugammadex.
Cada vial de 2 mL contiene sugammadex sódico equivalente a 200 mg de sugammadex. Cada vial de 5 mL contiene sugammadex sódico equivalente a 500 mg de sugammadex.
Excipiente(s) con efecto conocido
Contiene hasta 9,7 mg/mL de sodio (ver sección 4.4)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable (inyectable).
Solución transparente de incolora a ligeramente amarilla.
La solución tiene un pH entre 7 y 8 y una osmolaridad entre 300 y 500 mOsm/Kg.
4. DATOS CLÍNICOS
Reversión del bloqueo neuromuscular inducido por rocuronio o vecuronio en adultos.
Para la población pediátrica: sólo se recomienda el uso de sugammadex en niños y adolescentes, de edades comprendidas entre 2 y 17 años, para la reversión de rutina del bloqueo inducido por rocuronio.
Posología
Sugammadex sólo deberá ser administrado por, o bajo la supervisión de un anestesiólogo.
Se recomienda el uso de una técnica de monitoreo neuromuscular apropiada para monitorear la recuperación del bloqueo neuromuscular. (Ver sección 4.4.).
La dosis recomendada de sugammadex depende del nivel del bloqueo neuromuscular a revertir.
La dosis recomendada no depende de la pauta posológica de la anestesia aplicada.
Sugammadex puede ser utilizado para revertir diferentes niveles de bloqueo neuromuscular inducido por rocuronio o vecuronio:
Adultos:
Reversión de rutina:
Se recomienda una dosis de 4 mg/Kg de sugammadex si la recuperación ha alcanzado por lo menos 1-2 respuestas del contaje postetánico (PTC) tras el bloqueo inducido con rocuronio o vecuronio. El tiempo medio para recuperar el ratio T4/T1 a 0,9 es alrededor de 3 minutos (ver sección 5.1).
Se recomienda la administración de una dosis de 2 mg/Kg de sugammadex si se ha producido recuperación espontánea hasta al menos la reaparición de T2 tras el bloqueo inducido por rocuronio o vecuronio. El tiempo medio para recuperar el ratio T4/T1 a 0,9 es de alrededor de 2 minutos (ver sección 5.1).
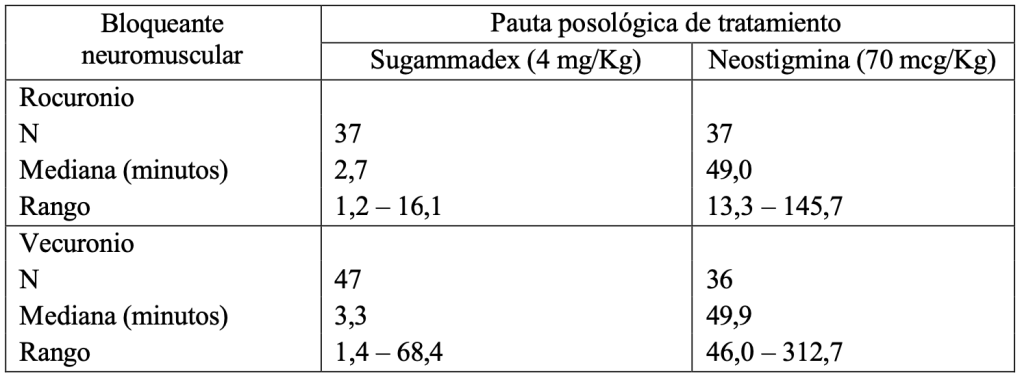
Si se utilizan las dosis recomendadas para la reversión de rutina, el tiempo medio para recuperar el ratio T4/T1 a 0,9 para rocuronio será ligeramente más rápido comparado con el bloqueo neuromuscular inducido por vecuronio (ver sección 5.1).
Reversión inmediata del bloqueo neuromuscular inducido por rocuronio:
Si hay una necesidad clínica de reversión inmediata tras la administración de rocuronio, se recomienda administrar una dosis de 16 mg/Kg de sugammadex. Si se administran 16 mg/Kg de sugammadex 3 minutos después de una dosis en bolus de 1,2 mg/Kg de bromuro de rocuronio, se puede esperar la recuperación del ratio T4/T1 a 0,9 en un tiempo medio de 1,5 minutos aproximadamente (ver sección 5.1).
No existen datos disponibles para recomendar el uso de sugammadex en la reversión inmediata tras el bloqueo inducido por vecuronio.
Repetición de la dosis de sugammadex:
En el caso excepcional de que se volviera a producir un bloqueo neuromuscular posoperatorio (ver sección 4.4) después de la administración de una dosis inicial de 2 mg/Kg o 4 mg/Kg de sugammadex, se recomienda administrar otra dosis de 4 mg/Kg de sugammadex. Después de la segunda dosis de sugammadex, se deberá monitorizar estrechamente al paciente, para comprobar la recuperación sostenida de la funcionalidad neuromuscular.
Repetición de la dosis de rocuronio o vecuronio después del tratamiento con sugammadex:
Para tiempos de espera para la readministración de rocuronio o vecuronio tras la reversión con sugammadex, ver sección 4.4.
Información adicional sobre poblaciones especiales
Insuficiencia renal:
No se recomienda el uso de sugammadex en pacientes con insuficiencia renal grave (incluyendo pacientes que requieren diálisis (ClCr < 30 mL/min)) (ver sección 4.4.).
Ensayos en pacientes con insuficiencia renal grave no han proporcionado información de seguridad suficiente para apoyar el uso de sugammadex en estos pacientes (ver también sección 5.1).
Insuficiencia renal leve y moderada (aclaramiento de creatinina ≥ 30 y < 80 mL/min): las recomendaciones de dosis son las mismas que para los adultos sin insuficiencia renal.
Pacientes de edad avanzada:
Tras la administración de sugammadex cuando reaparece el T2 tras el bloqueo inducido por rocuronio, el tiempo medio para recuperar el ratio T4/T1 a 0,9 en adultos (18-64 años) fue 2,2 minutos, en adultos de edad avanzada (65-74 años) fue 2,6 minutos y en adultos ancianos (a partir de 75 años) fue de 3,6 minutos. Aunque los tiempos de recuperación en personas de edad avanzada tienden a ser más lentos, se deben seguir las mismas recomendaciones posológicas que las indicadas para adultos (ver sección 4.4).
Pacientes obesos:
En pacientes obesos, incluidos pacientes con obesidad mórbida (índice de masa corporal ≥ 40 Kg/m2), la dosis de sugammadex se debe basar en el peso corporal real. Se deben seguir las mismas recomendaciones posológicas que las indicadas para los adultos.
Insuficiencia hepática:
No se han realizado estudios en pacientes con insuficiencia hepática. Se debe tener precaución cuando se considere el uso de sugammadex en pacientes con insuficiencia hepática grave o insuficiencia hepática acompañada de coagulopatía (ver sección 4.4).
Insuficiencia hepática de leve a moderada: no se requieren ajustes de dosis porque sugammadex se elimina principalmente por vía renal.
Población pediátrica
Reversión de rutina:
Se recomienda la administración de una dosis de 4 mg/kg de sugammadex para la reversión del bloqueo inducido por rocuronio si la recuperación ha alcanzado al menos 1-2 PTC.
Se recomienda la administración de una dosis de 2 mg/kg para la reversión del bloqueo inducido por rocuronio cuando reaparece el T2 (ver sección 5.1).
Reversión inmediata:
No se ha investigado la reversión inmediata en niños y adolescentes.
Recién nacidos a término (neonatos) y niños pequeños:
La experiencia con el uso de sugammadex en niños pequeños (de 30 días a 2 años) es limitada y no se ha estudiado en neonatos (menos de 30 días). No se recomienda el uso de sugammadex en recién nacidos a término y niños pequeños hasta que se disponga de más datos.
Forma de administración
Sugammadex se debe administrar por vía intravenosa en una única inyección en bolus. La inyección en bolus se debe administrar rápidamente, en un intervalo de 10 segundos, en una vía intravenosa preexistente (ver sección 6.6). En los ensayos clínicos, sugammadex sólo se ha administrado en forma de inyección única en bolus.
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Tal como es habitual en la práctica posanestesia tras el bloqueo neuromuscular, se recomienda controlar al paciente en el posoperatorio inmediato para detectar efectos inesperados como la reaparición del bloqueo neuromuscular.
Monitorización de la función respiratoria durante la recuperación:
Es obligatorio aplicar ventilación mecánica a los pacientes hasta que se recupere la respiración espontánea de forma adecuada tras la reversión del bloqueo neuromuscular. Incluso si la recuperación del bloqueo neuromuscular fuera completa, el resto de los medicamentos que se utilizan en el periodo peri y posoperatorio pueden deprimir la función respiratoria, por lo que puede continuar siendo necesaria la aplicación de ventilación mecánica.
Si el bloqueo neuromuscular se vuelve a producir tras la extubación, se debe proporcionar ventilación adecuada.
Reaparición del bloqueo neuromuscular:
En ensayos clínicos con pacientes tratados con rocuronio o vecuronio, donde se administró sugammadex utilizando una dosis establecida para la profundidad del bloqueo neuromuscular, se observó una incidencia de un 0,20% para la reaparición del bloqueo neuromuscular, basándose en la monitorización neuromuscular o en la evidencia clínica. El uso de dosis más bajas que las recomendadas puede producir un riesgo mayor de reaparición del bloqueo neuromuscular después de la reversión inicial, por lo que no se recomienda (ver sección 4.2 y sección 4.8).
Efecto sobre la hemostasia:
En un estudio en pacientes voluntarios, dosis de 4 mg/Kg y 16 mg/Kg de sugammadex dieron lugar a prolongaciones medias máximas del tiempo parcial de tromboplastina activada (aPTT) en un 17 y un 22%, respectivamente, y cociente internacional normalizado del tiempo de protrombina [PT(INR)] en un 11 y 22%, respectivamente. Estas prolongaciones medias en la aPTT y PT (INR) fueron de corta duración (≤ 30 minutos). Basándose en la base de datos clínicos (N=3.519), y en un estudio específico en 1.184 pacientes sometidos a cirugía de fractura de cadera/cirugía mayor de reemplazo articular, no hubo efecto clínicamente relevante en la incidencia de complicaciones de hemorragias peri y posoperatorias con sugammadex 4 mg/Kg solo o en combinación con anticoagulantes.
En experimentos in vitro se observó una interacción farmacodinámica (prolongación del tiempo parcial de tromboplastina activada [aPTT] y del tiempo de protrombina [PT]) con antagonistas de la vitamina K, heparina no fraccionada, heparinoides de bajo peso molecular, rivaroxaban y dabigatran. En pacientes que reciben anticoagulación profiláctica posoperatoria habitual, esta interacción farmacodinámica no es clínicamente relevante. Se debe actuar con precaución cuando se considere la utilización de sugammadex en pacientes que reciben tratamiento anticoagulante para una enfermedad preexistente o concomitante.
No se puede descartar un incremento del riesgo de hemorragias en pacientes:
• con deficiencias hereditarias de factores de la coagulación dependientes de la vitamina K;
• con coagulopatías preexistentes;
• tratados con derivados cumarínicos y con un factor INR por encima de 3,5;
• que utilicen anticoagulantes y que reciban una dosis de 16 mg/kg de sugammadex.
Si existe necesidad médica de administrar sugammadex a estos pacientes, el anestesiólogo decidirá si los beneficios superan el posible riesgo de complicaciones hemorrágicas, teniendo en consideración los antecedentes de episodios hemorrágicos de los pacientes y el tipo de cirugía programada. Se recomienda controlar la hemostasia y los parámetros de coagulación si se administra sugammadex a estos pacientes.
Tiempos de espera recomendados para volver a administrar los bloqueantes neuromusculares tras la reversión con sugammadex:
Tabla 1: Readministración de rocuronio o vecuronio tras la reversión de rutina (hasta 4 mg/Kg sugammadex):

Después de la readministración de 1,2 mg/Kg de rocuronio en 30 minutos tras la administración de sugammadex, el inicio del bloqueo neuromuscular se puede prolongar hasta 4 minutos aproximadamente, y la duración del bloqueo neuromuscular se puede reducir hasta 15 minutos aproximadamente.
En base al modelo farmacocinético (PK), el tiempo de espera recomendado en pacientes con insuficiencia renal leve o moderada para la readministración de 0,6 mg/Kg de rocuronio o 0,1 mg/Kg de vecuronio tras la reversión de rutina con sugammadex, debe de ser de 24 horas. Si se requiere un tiempo de espera más corto, la dosis de rocuronio para un nuevo bloqueo neuromuscular debe de ser 1,2 mg/Kg.
Readministración de rocuronio o vecuronio tras la reversión inmediata (16 mg/Kg de sugammadex): En casos muy raros en los que se pueda requerir, se recomienda un tiempo de espera de 24 horas.
Si se necesitara administrar un bloqueo neuromuscular antes del tiempo de espera recomendado, se debe utilizar un bloqueante neuromuscular no esteroideo. El comienzo de un bloqueante neuromuscular despolarizante puede ser más lento de lo esperado debido a que, una fracción sustancial de los receptores nicotínicos postsinápticos pueden estar ocupados todavía por el bloqueante neuromuscular.
Insuficiencia renal:
No se recomienda el uso de sugammadex en pacientes con insuficiencia renal grave, incluidos los que requieren diálisis (ver sección 5.1).
Anestesia superficial:
En los ensayos clínicos, en los casos en los que se revirtió de forma intencionada el bloqueo neuromuscular durante la anestesia, se observaron ocasionalmente signos de anestesia superficial (movimientos, tos, espasmos faciales y succión del tubo endotraqueal). Si se revierte el bloqueo neuromuscular mientras se continúa con la anestesia, se deben administrar otras dosis de anestésico y/u opioide en la forma que esté indicada clínicamente.
Bradicardia acusada:
En casos raros, se ha observado bradicardia acusada pocos minutos después de la administración de sugammadex para la reversión del bloqueo neuromuscular. En ocasiones, la bradicardia puede producir una parada cardiaca. (Ver sección 4.8). Se debe monitorizar estrechamente a los pacientes para evitar cambios hemodinámicos durante y después de la reversión del bloqueo neuromuscular. Si se observa bradicardia clínicamente significativa, se debe administrar un tratamiento con anti-colinérgicos, tal como atropina.
Insuficiencia hepática:
Sugammadex no se metaboliza ni se elimina por el hígado; por tanto, no se han realizado estudios específicos en pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática grave se deben tratar con gran precaución. En caso de que la insuficiencia hepática se acompañe de coagulopatía ver la información del efecto sobre la hemostasia.
Uso en la Unidad de Cuidados Intensivos (UCI):
No se ha investigado sugammadex en pacientes que han recibido rocuronio o vecuronio en la UCI.
Reversión del bloqueo neuromuscular de otros medicamentos que no sean rocuronio o vecuronio:
El tratamiento con sugammadex no se debe utilizar para revertir el bloqueo inducido por bloqueantes neuromusculares no esteroideos tales como la succinilcolina o los derivados benzilisoquinólicos.
El tratamiento con sugammadex no se debe utilizar para la reversión del bloqueo neuromuscular inducido por bloqueantes neuromusculares esteroideos que no sean el rocuronio o el vecuronio, ya que no se dispone de datos de eficacia y seguridad en estos casos. Se dispone de datos limitados acerca de la reversión del bloqueo inducido por pancuronio, pero no se recomienda utilizar sugammadex en esta situación.
Retraso de la recuperación:
Situaciones asociadas con un tiempo de circulación prolongado, tal como cardiopatías, edad avanzada (ver sección 4.2 sobre el tiempo de recuperación en pacientes de edad avanzada), o estados edematosos (por ejemplo, insuficiencia hepática grave), se pueden asociar con tiempos de recuperación más prolongados.
Reacciones de hipersensibilidad al medicamento:
Los médicos deben estar preparados para la posibilidad de que se produzcan reacciones de hipersensibilidad (que incluyen reacciones anafilácticas) y deben tomar las precauciones necesarias (ver sección 4.8).
Sodio:
Este medicamento contiene hasta 9,7 mg de sodio por mL, equivalente a 0,5% de la ingesta máxima diaria de 2 g de sodio recomendada por la OMS para un adulto.
La información de esta sección se basa en la afinidad de la unión entre el sugammadex y otros medicamentos, en los experimentos no-clínicos, en ensayos clínicos y en simulaciones con un modelo que tiene en cuenta el efecto farmacodinámico de los bloqueantes neuromusculares y la interacción farmacocinética entre los bloqueantes neuromusculares y sugammadex. En base a estos datos, no se espera que se produzcan interacciones farmacodinámicas clínicamente significativas con otros medicamentos, exceptuando los siguientes:
Toremifeno y ácido fusídico: no se puede excluir la posibilidad de que se produzcan interacciones por desplazamiento (no se esperan interacciones de la captura de relevancia clínica).
Anticonceptivos hormonales: No se puede excluir la posibilidad de que se produzca una interacción de la captura de relevancia clínica (no se esperan interacciones por desplazamiento).
Interacciones que afectan potencialmente a la eficacia de sugammadex (interacciones por desplazamiento): Teóricamente, la administración de ciertos medicamentos después del tratamiento con sugammadex, puede producir un desplazamiento del rocuronio o el vecuronio del complejo de sugammadex y en consecuencia, se puede observar una reaparición del bloqueo neuromuscular. En esta situación, se debe administrar al paciente ventilación mecánica. Se debe suspender la administración del medicamento que causa el desplazamiento si se administra por perfusión. En situaciones en las que se puedan anticipar interacciones potenciales por desplazamiento por la administración parenteral de otro medicamento en un periodo de 7,5 horas tras la administración de sugammadex, se debe monitorizar cuidadosamente a los pacientes para detectar los signos de reaparición de bloqueo neuromuscular (hasta 15 minutos aproximadamente).
Toremifeno:
En el caso de la administración concomitante con toremifeno, que posee una afinidad de unión relativamente alta por el sugammadex y para el cual pueden estar presentes concentraciones plasmáticas relativamente elevadas, se puede producir cierto desplazamiento del rocuronio o vecuronio del complejo con sugammadex. Los médicos deben de ser conscientes de que la recuperación del ratio T4/T1 a 0,9 se puede por tanto retrasar en pacientes que han recibido toremifeno en el mismo día de la intervención quirúrgica.
Administración intravenosa de ácido fusídico:
El uso de ácido fusídico en la fase preoperatoria puede producir cierto retraso en la recuperación del ratio T4/T1 a 0,9. No se espera reaparición del bloqueo neuromuscular en la fase posoperatoria, ya que la perfusión del ácido fusídico dura varias horas y los niveles en sangre se acumulan más de 2-3 días.
Ver sección 4.2 para volver a administrar sugammadex.
Interacciones que afectan potencialmente a la eficacia de otros medicamentos (interacciones de la captura): La administración de sugammadex puede producir la disminución de las concentraciones plasmáticas (libres) de ciertos medicamentos, por lo que la eficacia de los mismos puede disminuir. Si se observa esta situación, el médico deberá considerar volver a administrar el mismo medicamento, administrar un medicamento terapéuticamente equivalente (preferiblemente que pertenezca a una clase química distinta) y/o aplicar las intervenciones no farmacológicas que sean necesarias.
Anticonceptivos hormonales:
Se prevé que la interacción entre sugammadex 4 mg/Kg y el progestágeno produzca una disminución en la exposición al progestágeno (34% de la AUC), similar a la disminución que se observa si una dosis diaria de un anticonceptivo oral se toma con 12 horas de retraso, lo que puede conducir a una reducción de la efectividad. En el caso de los estrógenos, se espera que el efecto sea inferior. Por tanto, la administración de una dosis en bolus de sugammadex se considera equivalente al olvido de una dosis diaria de un anticonceptivo esteroideo oral (ya sea combinado o con sólo progestágeno). Si el sugammadex se administra el mismo día que un anticonceptivo oral se debe referir a las recomendaciones en caso de olvido de una dosis del prospecto del anticonceptivo oral. En caso de anticonceptivos hormonales no orales, la paciente debe utilizar un anticonceptivo complementario no hormonal durante los siguientes 7 días y seguir las recomendaciones del prospecto del producto.
Interacciones debidas a la duración prolongada del efecto de rocuronio o vecuronio:
Si se utilizan medicamentos que potencian el bloqueo neuromuscular en el periodo posoperatorio, se debe prestar una especial atención a la posibilidad de que se produzca una reaparición del bloqueo neuromuscular. Ver los prospectos de rocuronio o vecuronio en los que se proporciona una lista de los medicamentos concretos que potencian el bloqueo neuromuscular. En caso de reaparición del bloqueo neuromuscular, el paciente puede requerir ventilación mecánica y repetición de la dosis de sugammadex (ver sección 4.2).
Interferencia con pruebas de laboratorio:
En general sugammadex no interfiere con las pruebas de laboratorio, con la posible excepción de la determinación de la progesterona en suero. Se observa interferencia con esta prueba a concentraciones de 100 microgramos/mL de sugammadex en plasma (nivel máximo de plasma tras 8 mg/Kg de inyección en bolus).
En un estudio en voluntarios, dosis de 4 mg/Kg y de 16 mg/Kg de sugammadex dieron lugar a prolongaciones medias máximas del aPTT en un 17 y un 22%, respectivamente, y del PT (INR) en un 11 y 22%, respectivamente. Estas prolongaciones medias en la aPTT y PT (INR) fueron de corta duración (≤ 30 minutos).
En experimentos in vitro se observó una interacción farmacodinámica (prolongación del tiempo parcial de tromboplastina activada [aPTT] y del tiempo de protrombina [PT]) con antagonistas de la vitamina K, heparina no fraccionada, heparinoides de bajo peso molecular, rivaroxaban y dabigatran (ver sección 4.4).
Población pediátrica
BRIDION
No se han realizado estudios de interacciones. Las interacciones que se mencionan anteriormente para los adultos, así como las advertencias incluidas en la sección 4.4, se deben tener también en cuenta para la población pediátrica.
Embarazo
No existen datos clínicos sobre la exposición de embarazadas a sugammadex.
Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos sobre el embarazo, desarrollo embriofetal, parto o desarrollo posnatal.
Se debe actuar con precaución cuando se administre sugammadex a mujeres embarazadas.
Lactancia
Se desconoce si sugammadex se excreta en la leche materna humana. En estudios en animales se ha observado que sugammadex se excreta en la leche materna. La absorción oral de ciclodextrinas es por lo general baja y no se prevé que tenga efecto sobre el lactante tras la administración de una dosis única a la mujer durante el periodo de lactancia.
Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento, tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No se han investigado los efectos de sugammadex en la fertilidad humana. Estudios en animales para evaluar la fertilidad no muestran efectos nocivos.
BRIDION no tiene influencia conocida sobre la capacidad para conducir y utilizar máquinas.
Resumen del perfil de seguridad
BRIDION se administró de forma concomitante con bloqueantes neuromusculares y anestésicos en pacientes quirúrgicos. La causalidad de los efectos adversos es por lo tanto difícil de evaluar.
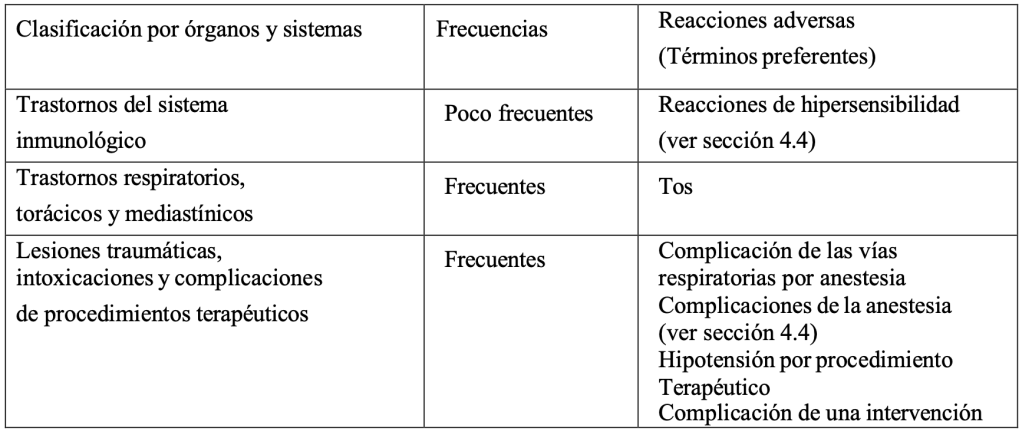
Las reacciones adversas notificadas más frecuentemente en pacientes quirúrgicos fueron tos, complicación de las vías respiratorias por anestesia, complicaciones de la anestesia, hipotensión por procedimiento terapéutico y complicación de una intervención (Frecuentes (≥ 1/100 a < 1/10)).
Tabla 2: Tabla de reacciones adversas
La seguridad de sugammadex se ha evaluado en 3.519 pacientes únicos a través de una base de datos conjunta de seguridad fase I-III. Se notificaron las siguientes reacciones adversas en los ensayos controlados con placebo en los que los pacientes recibieron anestesia y/o bloqueantes neuromusculares (1.078 pacientes expuestos a sugammadex frente a 544 expuestos a placebo): [Muy frecuentes (≥1/10), frecuentes ≥1/100 a <1/10), poco frecuentes (≥1/1.000 a <1/100), raras (≥1/10.000 a <1/1.000), muy raras (<1/10.000)]
Descripción de las reacciones adversas seleccionadas
Reacciones de hipersensibilidad:
Se han producido reacciones de hipersensibilidad, incluyendo anafilaxia, en algunos pacientes y voluntarios (para obtener información sobre los voluntarios, ver más adelante Información sobre voluntarios sanos). En ensayos clínicos de pacientes quirúrgicos, estas reacciones fueron notificadas poco frecuentemente, y en los informes poscomercialización la frecuencia es desconocida.
Estas reacciones variaron de reacciones cutáneas aisladas a reacciones sistémicas graves (esto es, anafilaxia, shock anafiláctico) y han tenido lugar en pacientes sin exposición previa a sugammadex.
Los síntomas asociados a estas reacciones pueden incluir: rubefacción, urticaria, erupción eritematosa, hipotensión (grave), taquicardia, hinchazón de lengua, hinchazón de faringe, broncoespasmo y acontecimientos pulmonares obstructivos. Las reacciones de hipersensibilidad graves pueden ser mortales.
Complicación de las vías respiratorias por anestesia:
Las complicaciones de las vías respiratorias por anestesia, incluyeron espasmos relacionados con el final de la anestesia o con la extubación contra el tubo endotraqueal, tos, leves espasmos relacionados con el final de la anestesia o con la extubación, reacción de despertar durante la cirugía, tos durante el procedimiento anestésico o durante la cirugía, o respiración espontánea del paciente relacionada con el procedimiento anestésico.
Complicación de la anestesia:
Las complicaciones de la anestesia, que indican recuperación de la función neuromuscular, incluyen movimiento de una extremidad o del cuerpo o tos durante la administración de la anestesia o durante la cirugía, espasmos faciales o succión en el tubo endotraqueal. Ver sección 4.4 anestesia superficial.
Complicación de una intervención:
Las complicaciones de una intervención incluyeron tos, movimientos, taquicardia, bradicardia y el aumento de la frecuencia cardiaca.
Bradicardia acusada:
Tras la comercialización, se han observado casos aislados de bradicardia acusada y bradicardia con parada cardíaca pocos minutos después de la administración de sugammadex (ver sección 4.4).
Reaparición del bloqueo neuromuscular:
En ensayos clínicos con pacientes tratados con rocuronio o vecuronio, donde se administró sugammadex utilizando una dosis establecida para la profundidad del bloqueo neuromuscular (N=2.022), se observó una incidencia de un 0,20% para la reaparición del bloqueo neuromuscular, basándose en la monitorización neuromuscular o en la evidencia clínica (ver sección 4.4).
Información sobre voluntarios sanos:
Un estudio aleatorizado y doble ciego, evaluó la incidencia de reacciones de hipersensibilidad al medicamento en voluntarios sanos que recibieron hasta 3 dosis de placebo (N=76), de sugammadex 4 mg/Kg (N=151) o de sugammadex 16 mg/Kg (N=148). Las notificaciones de sospecha de hipersensibilidad se establecieron por una comisión independiente. La incidencia de hipersensibilidad establecida fue de un 1,3%, de un 6,6% y de un 9,5% en los grupos placebo, en los de sugammadex 4 mg/Kg y en los de sugammadex 16 mg/Kg, respectivamente. No hubo notificaciones de anafilaxia después de la administración de placebo o de sugammadex 4 mg/Kg. Solo hubo un caso de anafilaxia establecida después de la administración de la primera dosis de sugammadex 16 mg/Kg (incidencia de un 0,7%). Al repetir la dosis de sugammadex, no hubo indicios de aumento de la frecuencia o de la gravedad de la hipersensibilidad.
En un estudio anterior de diseño similar, hubo tres casos de anafilaxia establecida, todos después de la administración de sugammadex 16 mg/Kg (incidencia de un 2,0%).
En la base de datos conjunta de los ensayos en fase I, las reacciones adversas frecuentes (≥ 1/100 a < 1/10), o muy frecuentes (≥ 1/10) y más frecuentes entre los pacientes tratados con sugammadex que en los del grupo placebo, incluyen disgeusia (10,1%), cefalea (6,7%), náuseas (5,6%), urticaria (1,7%), prurito (1,7%), mareo (1,6%), vómitos (1,2%) y dolor abdominal (1,0%).
Información adicional para poblaciones especiales
Complicaciones pulmonares:
En datos de poscomercialización y en un ensayo clínico específico, en pacientes con antecedentes de complicaciones pulmonares, se notificó broncoespasmo como reacción adversa posiblemente relacionada con el tratamiento. Al igual que con todos los pacientes con antecedentes de complicaciones pulmonares el médico debe estar atento a la posible aparición de broncoespasmo.
Población pediátrica
En los estudios de pacientes pediátricos entre 2 y 17 años, el perfil de seguridad de sugammadex (hasta 4 mg/kg) fue en general, similar al perfil observado en adultos.
Pacientes con obesidad mórbida
En un ensayo clínico específico en pacientes con obesidad mórbida, el perfil de seguridad fue en general, similar al perfil en pacientes adultos en ensayos combinados de Fase 1 a la 3 (ver Tabla 2).
Pacientes con enfermedad sistémica grave
En un ensayo en pacientes que fueron evaluados como Clase 3 o 4 según la Sociedad Americana de Anestesiología (ASA, por sus siglas en inglés) (es decir, pacientes con enfermedad sistémica grave o pacientes con enfermedad sistémica grave que constituye una constante amenaza a la vida), el perfil de reacciones adversas en estos pacientes Clase ASA 3 y 4 fue en general similar al de pacientes adultos en ensayos combinados de Fase 1 a 3 (ver Tabla 2). Ver sección 5.1.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación.
Durante los ensayos clínicos, se notificó un caso de sobredosis accidental con 40 mg/Kg sin que se produjera ninguna reacción adversa significativa. En estudios de tolerancia en humanos sugammadex se administró en dosis de hasta 96 mg/Kg. No se notificaron reacciones adversas relacionadas con la dosis ni reacciones adversas graves.
Sugammadex se puede eliminar mediante hemodiálisis con un filtro de alto flujo, pero no con un filtro de bajo flujo. Los ensayos clínicos indican que las concentraciones de sugammadex en el plasma se reducen hasta en un 70% después de una sesión de diálisis de 3 a 6 horas.
5. PROPIEDADES FARMACOLÓGICAS
Grupo farmacoterapéutico: todos los demás grupos terapéuticos; antídotos, código ATC: V03AB35
Mecanismo de acción:
Sugammadex es una gamma ciclodextrina modificada, que actúa como un Agente Selectivo de Unión a Bloqueantes (Selective Relaxant Binding Agent). Forma un complejo con los bloqueantes neuromusculares rocuronio o vecuronio en plasma y por tanto reduce la cantidad de bloqueante neuromuscular disponible para unirse a los receptores nicotínicos en la unión neuromuscular. Esto produce una reversión del bloqueo neuromuscular inducido por el rocuronio o el vecuronio.
Efectos farmacodinámicos:
Sugammadex se ha administrado en dosis desde 0,5 mg/Kg a 16 mg/Kg en estudios de dosis-respuesta del bloqueo inducido por rocuronio (0,6; 0,9; 1,0 y 1,2 mg/Kg de bromuro de rocuronio con y sin dosis de mantenimiento) y el bloqueo inducido por vecuronio (0,1 mg/kg de bromuro de vecuronio con y sin dosis de mantenimiento), a diferentes puntos temporales/profundidad del bloqueo. En estos estudios se observó una clara relación dosis-respuesta.
Eficacia clínica y seguridad:
Sugammadex se puede administrar a distintos tiempos tras la administración de rocuronio o vecuronio:
Reversión de rutina – bloqueo neuromuscular profundo:
En un ensayo pivotal los pacientes se asignaron aleatoriamente al grupo de rocuronio o vecuronio. Tras la última dosis de rocuronio o vecuronio, a 1-2 PTCs, se administraron 4 mg/Kg de sugammadex o 70 mcg/Kg de neostigmina de forma aleatoria. El tiempo desde el inicio de la administración de sugammadex o neostigmina hasta la recuperación del ratio T4/T1 a 0,9 fue:
Tabla 3: Tiempo (minutos) desde la administración de sugammadex o neostigmina en bloqueo neuromuscular profundo (1-2 PTCs) tras rocuronio o vecuronio hasta la recuperación del ratio T4/T1 a 0,9
Reversión de rutina – bloqueo neuromuscular moderado:
En otro ensayo pivotal los pacientes se asignaron aleatoriamente al grupo de rocuronio o vecuronio. Tras la última dosis de rocuronio o vecuronio, cuando reapareció el T2, se administraron 2 mg/kg de sugammadex o 50 mcg/kg de neostigmina de forma aleatoria. El tiempo desde el inicio de la administración de sugammadex o neostigmina hasta la recuperación del ratio T4/T1 a 0,9 fue:
Tabla 4: Tiempo (minutos) desde la administración de sugammadex o neostigmina reaparición de T2 después de rocuronio o vecuronio hasta la recuperación de la relación T4/T1 a 0,9

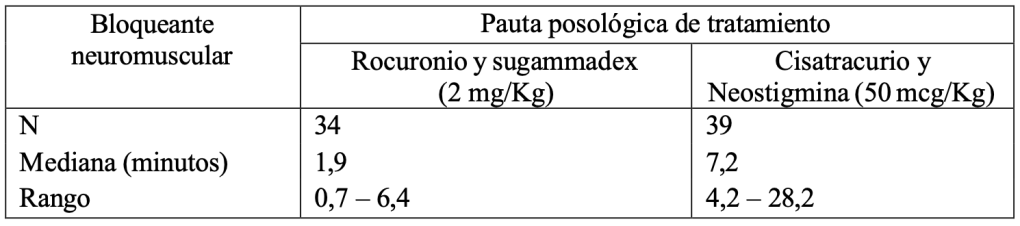
La reversión producida por sugammadex del bloqueo neuromuscular inducido por rocuronio se comparó con la reversión producida por neostigmina del bloqueo neuromuscular inducido por cisatracurio. Cuando reapareció T2 se administró una dosis de 2 mg/kg de sugammadex o 50 mcg/kg de neostigmina. La administración de sugammadex produjo una reversión del bloqueo neuromuscular inducido por rocuronio más rápida que la producida por neostigmina sobre el bloqueo neuromuscular inducido por cisatracurio:
Tabla 5: Tiempo (en minutos) desde la administración de sugammadex o neostigmina, cuando reapareció el T2 tras rocuronio o cisatracurio para la recuperación del ratio T4/T1 a 0,9
Reversion inmediata:
El tiempo de recuperación desde el bloqueo neuromuscular inducido por succinilcolina (1 mg/Kg) se comparó con la recuperación inducida por sugammadex (16 mg/Kg, 3 minutos después) – del bloqueo neuromuscular inducido por rocuronio (1,2 mg/Kg).
Tabla 6: Tiempo (minutos) desde la administración de rocuronio y sugammadex o succinilcolina para la recuperación de T1 al 10%

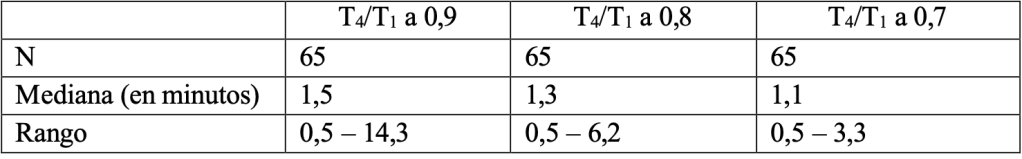
En un análisis combinado se obtuvieron los siguientes tiempos de recuperación para una dosis de sugammadex de 16 mg/kg tras 1,2 mg/Kg de bromuro de rocuronio:
Tabla 7: Tiempo (en minutos) desde la administración de sugammadex que se realizó a los 3 minutos tras la administración de rocuronio para la recuperación del ratio T4/T1 a 0,9, 0,8 o 0,7
Insuficiencia renal:
Se comparó la eficacia y seguridad de sugammadex en dos ensayos abiertos, en pacientes quirúrgicos con y sin insuficiencia renal grave. En un ensayo, sugammadex se administró tras el bloqueo inducido por rocuronio en 1-2 PTCs (4 mg/Kg; N=68); en el otro ensayo, se administró sugammadex en la reaparición de T2 (2 mg/Kg; N=30). La recuperación del bloqueo fue ligeramente más larga en pacientes con insuficiencia renal grave en relación con pacientes sin insuficiencia renal. En estos ensayos, en pacientes con insuficiencia renal grave no se notificó bloqueo neuromuscular residual ni reaparición de bloqueo neuromuscular.
Pacientes con obesidad mórbida:
En un ensayo de 188 pacientes diagnosticados con obesidad mórbida, se investigó el tiempo de recuperación del bloqueo neuromuscular moderado o profundo, inducido por rocuronio o vecuronio.
Los pacientes recibieron de forma aleatoria y doble ciego 2 mg/Kg o 4 mg/Kg de sugammadex, según lo adecuado para el nivel del bloqueo y tratados en función del peso corporal real o del peso corporal ideal.
Combinados en función de la profundidad del bloqueo y del bloqueante neuromuscular, el tiempo medio para recuperar el ratio del tren de cuatro (TOF) ≥ 0,9 en pacientes tratados según el peso corporal real (1,8 minutos) fue estadística y significativamente más rápido (p < 0,0001) en comparación con pacientes tratados según el peso corporal ideal (3,3 minutos).
Población pediátrica:
En un ensayo con 288 pacientes de edades comprendidas entre 2 y < 17 años, se investigó la seguridad y eficacia de sugammadex frente a neostigmina como un agente para la reversión del bloqueo neuromuscular inducido por rocuronio o vecuronio. La recuperación desde un bloqueo moderado hasta el ratio del TOF ≥ 0,9 fue significativamente más rápido en el grupo de sugammadex de 2 mg/kg comparado con el grupo de neostigmina (media geométrica de 1,6 minutos para sugammadex 2 mg/kg y 7,5 minutos para neostigmina, ratio de las medias geométricas de 0,22, IC del 95 % (0,16, 0,32), (p<0,0001)). Sugammadex 4 mg/kg alcanzó la reversión desde un bloqueo profundo con una media geométrica de 2,0 minutos, similar a los resultados observados en adultos. Estos efectos fueron consistentes para todos los grupos de edad estudiados (entre 2 y < 6; 6 y < 12; 12 y < 17 años) y tanto para rocuronio como para vecuronio. Ver sección 4.2.
Pacientes con enfermedad sistémica grave:
En un ensayo con 331 pacientes que fueron evaluados como Clase ASA 3 o 4 se investigó la incidencia de aparición de arritmias durante el tratamiento (bradicardia sinusal, taquicardia sinusal y otras arritmias cardiacas) tras la administración de sugammadex.
En los pacientes que recibieron sugammadex (2 mg/kg, 4 mg/kg o 16 mg/kg), la incidencia de aparición de arritmias durante el tratamiento fue en general similar a neostigmina (50 μg/kg hasta una dosis máxima de 5 mg) + glicopirrolato (10 μg/kg hasta una dosis máxima de 1 mg). El perfil de reacciones adversas en estos pacientes Clase ASA 3 y 4 fue en general similar al de pacientes adultos en ensayos combinados de Fase 1 a 3; por tanto no es necesario un ajuste de dosis. Ver sección 4.8.
Los parámetros farmacocinéticos de sugammadex se calcularon a partir de la suma total de las concentraciones de sugammadex no complejadas y las sí complejadas. Se espera que parámetros farmacocinéticos tales como el aclaramiento y el volumen de distribución sean los mismos para el sugammadex no complejado y el complejado en pacientes anestesiados.
Distribución:
El volumen de distribución de sugammadex observado en estado estacionario es de 11 a 14 litros aproximadamente en pacientes adultos con función renal normal (basado en un análisis farmacocinético convencional, no compartimental). Ni el sugammadex ni el complejo de sugammadex y rocuronio se unen a las proteínas plasmáticas ni a los eritrocitos, tal como se demostró in vitro utilizando plasma y sangre total de humanos varones. Sugammadex presenta una cinética lineal en el rango de dosificación de 1 a 16 mg/Kg, cuando se administra por vía intravenosa en bolus.
Metabolismo:
En los estudios preclínicos y clínicos no se observaron metabolitos de sugammadex y la única vía de eliminación observada fue la excreción renal del producto inalterado.
Eliminación:
En pacientes adultos anestesiados con función renal normal, la vida media de eliminación (t1/2) del sugammadex es de 2 horas aproximadamente, y el aclaramiento plasmático estimado es de 88 mL/min aproximadamente. Un estudio de balance de masas, demostró que > 90% de la dosis se excretaba antes de 24 horas. El 96% de la dosis fue excretado en orina, del que al menos, un 95% era sugammadex inalterado. La excreción en heces o en aire expirado fue del 0,02% de la dosis. La administración de sugammadex a voluntarios sanos produjo un aumento de la eliminación renal del complejo con rocuronio.
Poblaciones especiales:
Insuficiencia renal y edad:
En un estudio farmacocinético, comparando pacientes con insuficiencia renal grave con pacientes con función renal normal, los niveles de sugammadex en plasma fueron similares durante la primera hora tras la administración de la dosis y, posteriormente, los niveles disminuyeron más rápido en el grupo control. La exposición total al sugammadex se prolongó en pacientes con insuficiencia renal grave, siendo 17 veces mayor. Durante al menos 48 horas tras la administración de la dosis son detectables concentraciones bajas de sugammadex en pacientes con insuficiencia renal grave.
En un segundo estudio, comparando pacientes con insuficiencia renal grave o moderada con pacientes con función renal normal, el aclaramiento de sugammadex disminuyó progresivamente y la t1/2 se prolongó progresivamente con la disminución de la función renal. La exposición fue 2 y 5 veces mayor en pacientes con insuficiencia renal moderada y grave, respectivamente. Pasados 7 días tras la administración de la dosis, las concentraciones de sugammadex ya no eran detectables en pacientes con insuficiencia renal grave.
BRIDION
Tabla 8: A continuación, se presenta un resumen de los parámetros farmacocinéticos de sugammadex estratificados por edad y función renal:
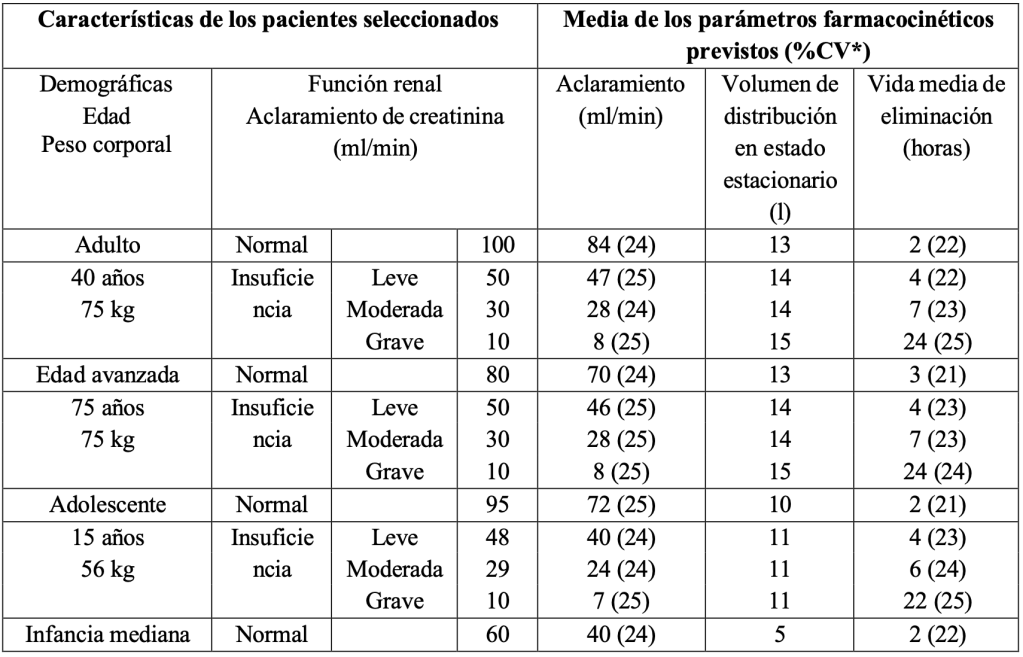
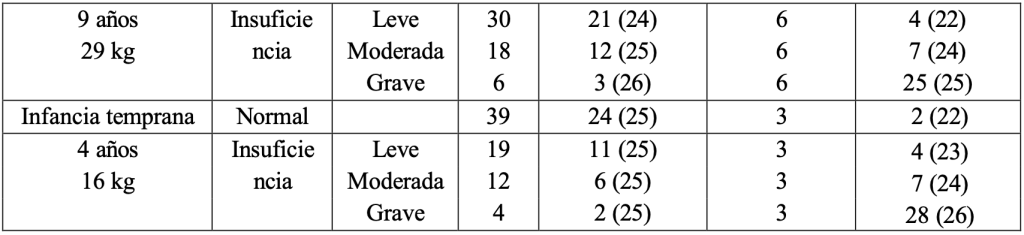
*CV=coeficiente de variación.
Sexo:
No se observaron diferencias en relación con el sexo.
Raza:
En un estudio en pacientes sanos japoneses y caucásicos no se observaron diferencias clínicamente relevantes de los parámetros farmacocinéticos. Existen datos limitados que no indican diferencias en los parámetros farmacocinéticos en pacientes de raza negra o afroamericanos.
Peso corporal:
El análisis de farmacocinética poblacional en adultos y pacientes de edad avanzada no mostró una relación clínicamente relevante entre el aclaramiento y el volumen de distribución con el peso corporal.
Obesidad:
En un ensayo clínico en pacientes con obesidad mórbida, se dosificó sugammadex 2 mg/Kg y 4 mg/Kg según el peso corporal real (n=76) o el peso corporal ideal (n=74). La exposición al sugammadex aumentó de forma lineal y dependiente de la dosis después de la administración según el peso corporal real o el peso corporal ideal. No se observaron diferencias clínicamente relevantes en los parámetros farmacocinéticos entre los pacientes con obesidad mórbida y la población general.
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción, tolerancia local y compatibilidad con la sangre. Sugammadex se elimina rápidamente en especies de animales en los estudios preclínicos, aunque se observó sugammadex residual en el tejido óseo y dental de ratas jóvenes. Los estudios preclínicos en ratas adultas, jóvenes y maduras demuestran que sugammadex no afecta negativamente al color de los dientes, ni a la calidad del hueso, ni a la estructura ósea, ni al metabolismo óseo. Sugammadex no tiene efectos sobre la reparación de fracturas ni en la remodelación del hueso.
6. DATOS FARMACÉUTICOS
Ácido clorhídrico 3,7% (para ajuste del pH) y/o hidróxido de sodio (para ajuste del pH) Agua para preparaciones inyectables.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6. Se ha comunicado incompatibilidad física con verapamilo, ondansetrón y ranitidina.
3 años.
Desde el punto de vista microbiológico, el producto diluido deberá ser utilizado de inmediato. Si no se lo utiliza de inmediato, los tiempos de almacenamiento durante el uso y las condiciones de almacenamiento previas al uso son responsabilidad del usuario y normalmente no superarían las 24 horas a una temperatura de 2 ̊C a 8 ̊C, a menos que la dilución se haya realizado en condiciones asépticas controladas y validadas.
Almacenar a una temperatura no mayor de 30 ̊C.
No congelar.
Conservar en el envase original para protegerlo de la luz.
Cuando no está protegido de la luz, el vial debe utilizarse dentro de 5 días.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
2 mL o 5 mL de solución en viales de vidrio tipo I cerrado con tapones de goma de clorobutilo con un precinto de aluminio.
Presentaciones: 10 viales de 2 mL o 10 viales de 5 mL.
Puede que solamente estén comercializados algunos tamaños de envases.
BRIDION se puede inyectar utilizando la misma vía que para una perfusión ya iniciada con las siguientes soluciones intravenosas: cloruro de sodio 9 mg/mL (0,9%), glucosa 50 mg/mL (5%), cloruro de sodio 4,5 mg/mL (0,45%) y glucosa 25 mg/mL (2,5%), solución Ringer lactato, solución Ringer y glucosa 50 mg/mL (5%) en cloruro de sodio 9 mg/mL (0,9%).
La vía de perfusión se debe lavar de forma adecuada (por ejemplo, con solución de cloruro de sodio al 0,9%) entre la administración de BRIDION y otros medicamentos.
Uso en la población pediátrica
Para pacientes pediátricos, BRIDION se puede diluir utilizando cloruro de sodio 9 mg/mL (0,9%) hasta una concentración de 10 mg/mL (ver sección 6.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
MERCK SHARP & DOHME (UK) LIMITED,
Reino Unido
8. FECHA DE REVISION DEL TEXTO
Febrero 2022
9. REFERENCIAS BIBLIOGRAFICAS
https://www.ema.europa.eu/en/documents/product-information/bridion-epar-product-information_es.pdf